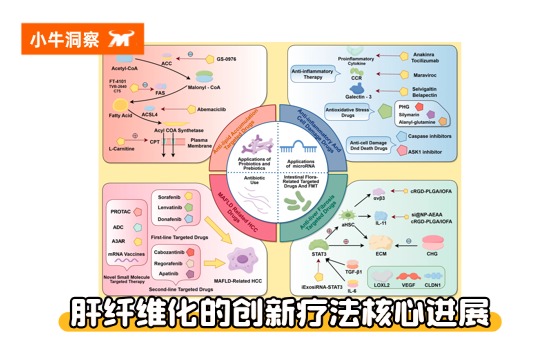
★ 上期我们主要探讨了肝纤维化的疾病认知与筛查的内容:小牛洞察 | 肝纤维化:被忽视的 “肝脏预警信号”,可逆但不容拖延
★ 本期聚焦肝纤维化的发病机制,深度解析以下核心要点:
1.详细介绍肝脏纤维化的病理过程:肝星状细胞(HSC)如何从”安静守护者”变成”肝纤维化肇事者”?
2.系统梳理肝纤维化相关的信号通路
3.纤维化逆转的核心机制是什么?为何早期干预如此重要?
肝纤维化的本质是“细胞外基质(ECM)过度沉积”,这个过程涉及肝脏内多种细胞的“跨界协作”和多条信号通路的“精准调控”,核心可概括为“损伤-炎症-活化-沉积”的四级级联反应。
PART 01
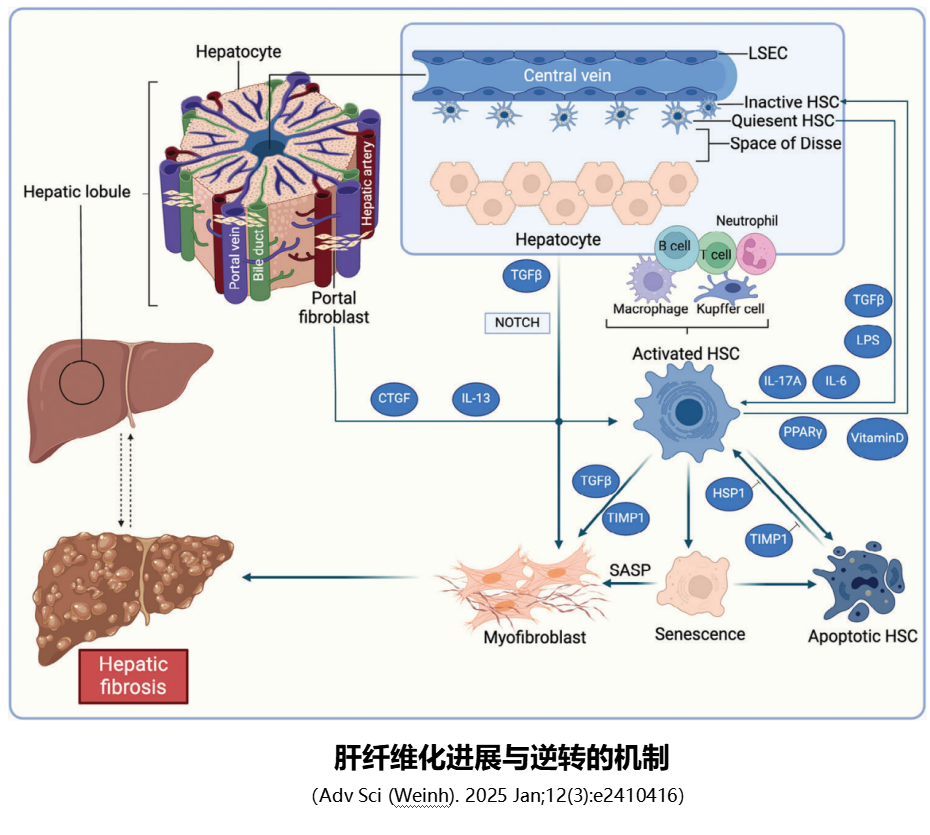
核心“肇事者”:肝星状细胞(HSC)的“叛变”之路
在肝脏的细胞群中,肝星状细胞(HSC)是纤维化的“核心执行者”,占肝脏细胞总数的5%~8%,正常状态下堪称肝脏的“安静守护者”:
- 静息态HSC:主要储存在肝脏的Disse间隙(肝血窦内皮细胞与肝细胞之间),负责储存维生素A,代谢脂质,此时不分泌胶原蛋白,对肝脏稳态至关重要;
- 活化态HSC:当肝脏受到损伤(病毒、酒精、脂肪毒性等),受损肝细胞会释放炎症因子(如TNF-α、TGF-β、IL-6/17A),同时Kupffer细胞(肝脏常驻巨噬细胞)被激活,释放大量促纤维化信号。这些信号会“唤醒”静息态HSC,使其转化为“肌成纤维细胞”——这是HSC的“叛变”关键。
活化后的HSC会发生三大变化:①高表达α-平滑肌肌动蛋白(α-SMA),具备收缩和增殖能力;②大量分泌胶原蛋白(尤其是Ⅰ型、Ⅲ型胶原)、纤维连接蛋白等细胞外基质,这些“疤痕材料”在肝脏内大量堆积;③分泌更多炎症因子和促纤维化因子,形成“炎症-活化”的恶性循环,加速纤维化进展。
- 值得注意的是,HSC并非肌成纤维细胞的唯一来源:门静脉成纤维细胞、骨髓来源的纤维细胞,以及肝细胞、胆管上皮细胞通过“上皮-间质转化(EMT)”,也能转化为肌成纤维细胞。这些细胞在衰老相关因子SASP(如结缔组织生长因子CTGF、IL-13、IL-11、IL-6、IL-1β、FGF2、TGF-β及COL1A1等)作用下向肌成纤维细胞转化。但HSC的贡献占比最高,是绝对核心。
- 当然。肝纤维化也可以逆转:刺激停止后,部分活化HSCs会凋亡,但TIMP1(金属蛋白酶组织抑制因子1)和HSP1(热休克蛋白1)会抑制这一凋亡过程;未凋亡的活化HSCs可在PPARγ和维生素D作用下失活。此外,淋巴细胞、库普弗细胞等免疫细胞在肝纤维化的进展和逆转中都有双向调节作用。
PART 02
关键“调控网”:驱动纤维化的核心信号通路
HSC的活化、增殖和细胞外基质的分泌,离不开多条信号通路的“指挥”,这些通路相互交织,形成复杂的调控网络:
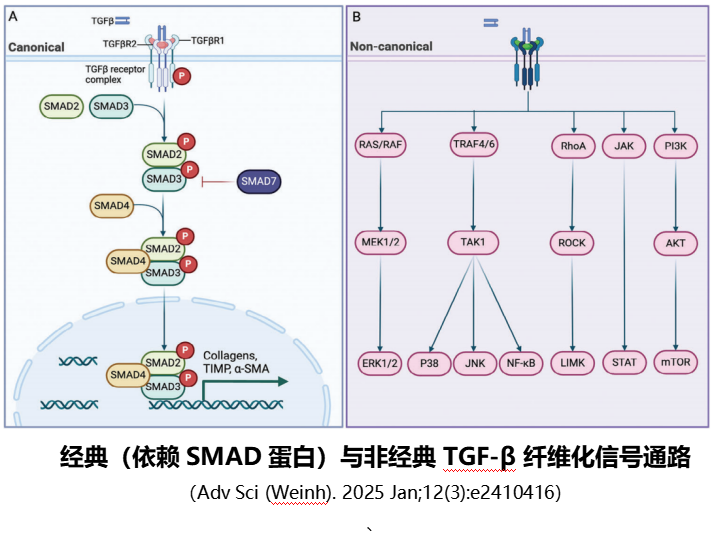
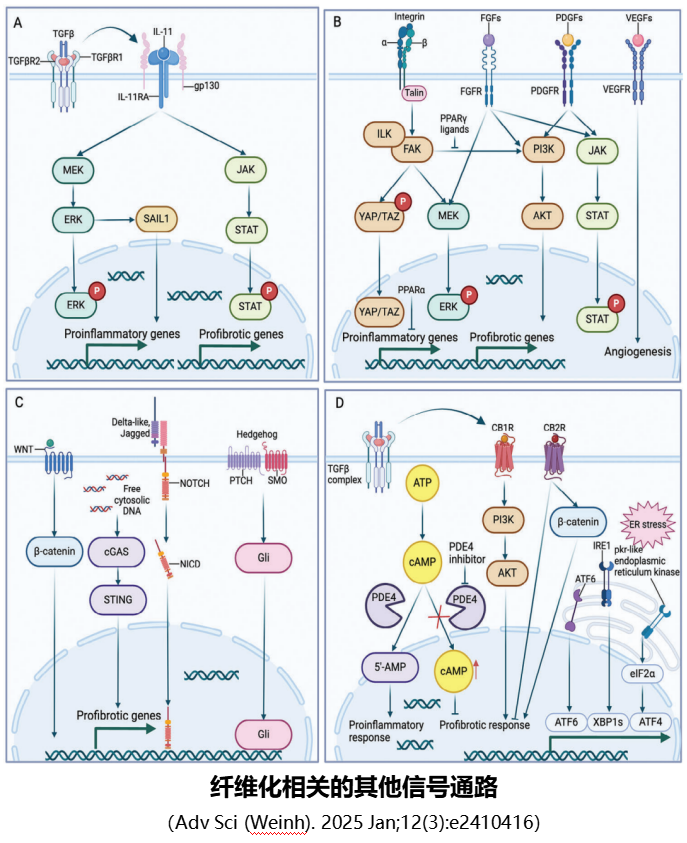
1.TGF-β/SMAD通路(核心中的核心):
这是纤维化最关键的信号通路,被称为“纤维化总指挥”。转化生长因子β(TGF-β)是主要的促纤维化细胞因子,当肝脏受损时,TGF-β大量释放,与HSC表面的受体结合后,激活下游的Smad2/3蛋白,这些蛋白进入细胞核后,启动胶原蛋白、α-SMA等基因的表达,直接促进HSC活化和细胞外基质合成。
同时,Smad7蛋白会对该通路进行负向调控,抑制纤维化进展,而在肝纤维化患者中,Smad7的表达往往降低,导致通路过度激活。此外,成熟的TGF-β也通过非经典信号通路发挥作用,即不依赖SMAD蛋白,通过激活其他信号通路(如RAS/RAF、TRAF4/6、RhoA/ROCK、JAK/STAT、PI3K/AKT等)发挥作用。这些通路涉及多种激酶和信号分子的级联反应,调控细胞增殖、分化、凋亡及细胞外基质代谢等过程,多层面参与纤维化相关的细胞生理和病理活动。
2.PDGF/PDGFR通路(增殖加速器):
血小板衍生生长因子(PDGF)是促进HSC增殖和迁移的最强因子。肝损伤后,血小板、巨噬细胞会释放PDGF,与HSC表面的PDGFR结合,通过激活MAPK、PI3K/AKT等下游信号,推动HSC快速增殖,同时增强其迁移能力,让活化的HSC在肝脏内广泛分布,扩大纤维化范围。
3.FGF/FGFR信号通路:
成纤维细胞生长因子(FGF)超家族含22种配体,其中FGF11-FGF14无法激活FGF受体,不属于经典 FGF 配体,而归为FGF同源因子(FHFs)。因此,哺乳动物中能激活受体的 18 种经典 FGF被分为6个亚家族:5个旁分泌亚家族和1个内分泌亚家族。
经典旁分泌亚家族通过先结合硫酸乙酰肝素(HS)或硫酸乙酰肝素蛋白聚糖(HSPG)辅助因子再结合FGF受体(FGFR1-4),非经典内分泌型FGF家族FGF19亚家族成员(FGF19、FGF21和FGF23)则为需要先结合组织特异性共受体Klotho蛋白,再与FGFR1-4结合。
FGF家族与受体结合后激活下游Ras/Raf/MEK/ERK-MAPK、PI3K/AKT、JAK/STAT等信号通路,参与肥胖、胰岛素抵抗、肿瘤、慢性肾脏病(CKD)等多种疾病。其中,敲低FGF2可通过AMP/ERK信号减少促炎性纤维化;FGF13敲除可通过调节微管稳定性预防血流动力学应激诱导的心脏纤维化。肝脏分泌的FGF21是循环FGF21的主要来源,是肝损伤的生物标志物及MAFLD或MASH的治疗靶点;在接受直接抗病毒治疗的丙肝患者中,FGF21升高与肝硬度呈负相关。
FGF19亚家族成员主要调节胆汁酸、胆固醇、脂肪酸、葡萄糖、矿物质(维生素D和磷酸盐等)平衡。
4.IL-11信号通路:
IL-11是IL-6细胞因子家族成员,在纤维化中起重要作用。IL-6家族具有一种广泛表达的关键细胞膜受体gp130(分子量130KD的糖蛋白),其与IL-11受体α亚基(IL-11RA)结合以实现信号转导。IL-6主要在免疫细胞中表达,而IL-11主要在基质细胞和上皮细胞中表达。
IL-11通过顺式或者反式作用于IL-11RA,在不同细胞类型中激活下游JAK/STAT3、MEK/ERK等信号通路。在酒精性肝病动物模型中,TGF-β激活HSC细胞ERK信号通路上调IL-11,引发肝纤维化,而阻断IL-11可以逆转肝纤维化。
5.代谢相关通路(代谢紊乱驱动者):
•PPAR家族通路:过氧化物酶体增殖物激活受体(PPAR)包括三种亚型:PPARα、PPARδ和PPARγ,控制着许多细胞内的代谢过程。PPARα主要调节脂肪酸的运输、酯化和氧化,其功能异常会导致脂肪在肝脏堆积,加重脂毒性损伤;PPARδ主要与脂肪酸的氧化和葡萄糖摄取有关;PPARγ主要调节脂肪细胞的分化、脂质储存和胰岛素敏感性。静息态HSC高表达PPARγ,能抑制其活化;而肝损伤后,PPARγ表达下降,HSC对脂质代谢的调控能力丧失,进而活化。
•FXR通路:法尼醇X受体(FXR)是胆汁酸代谢的关键受体,胆汁酸淤积会激活FXR,通过调节炎症因子和纤维化因子的表达,平衡肝脏的炎症与修复。FXR功能异常会导致胆汁酸代谢紊乱,加重肝损伤和纤维化。该通路的药物先行者以失败告终,但后续还有公司在研。
•THR-β通路:甲状腺激素受体(THR)属于核受体超家族,含THR-α(广泛分布,特别是心脏)和THR-β(主要集中在肝脏)两种亚型。THR-β在肝脏中参与脂质代谢和糖代谢,激活THR-β能减少肝脏甘油三酯,减少脂肪堆积,减轻脂毒性和炎症,进而抑制HSC活化。相当于从源头上解决纤维化问题。该通路已有药物成功上市。
6.其他关键通路:
•WNT/β-连环蛋白通路:该通路在胚胎发育中起重要作用,成年后基本沉默,而肝损伤后会被重新激活,通过促进HSC的糖酵解代谢,增强其增殖和活化能力,加速纤维化。
•Sonic Hedgehog信号通路:正常肝组织中无Hedgehog配体表达,肝损伤后HSC会分泌Shh(Sonic Hedgehog蛋白),诱导自身及周围细胞转分化为肌成纤维细胞;抑制Shh信号可显著抑制HSC活化和ECM合成,减轻肝纤维化。
•TLR4/NF-κB通路:肝脏受损后,肠道菌群紊乱导致脂多糖(LPS)入血,LPS与HSC表面的TLR4(Toll样受体4)结合,激活NF-κB通路,释放大量炎症因子(如TNF-α、IL-6),既加重肝脏炎症,又促进HSC活化。
•VEGF/VEGFR信号通路:长期暴露于环境污染物会诱导HSC产生VEGF,进而促进肝纤维化;VEGF通过调节肝脏血管生成和炎症微环境,间接参与纤维化进程。
•YAP/TAZ信号通路:YAP/TAZ(Yes相关蛋白/PDZ结合基序的转录共激活因子)激活后会促进HSC合成ECM,加重肝纤维化;在动物模型中,抑制YAP/TAZ与TEAD(转录增强相关结合域)的相互作用,可显著减轻肝脏ECM沉积和纤维化程度。
•ER应激信号通路:肝脏长期损伤会引发ER(内质网)应激,通过诱导肝细胞凋亡、HSC活化和炎症反应,推动肝纤维化;在MASH中,ER应激还会通过脂质代谢紊乱加重纤维化。
•cGAS/STING信号通路:肝细胞氧化应激介导的铁死亡会激活巨噬细胞的cGAS/STING(环状GMP-AMP合成酶/干扰素基因刺激因子)信号,进而引发肝损伤和纤维化;该通路还能促进肝窦微血管血栓形成,加重肝脏微循环障碍和纤维化。
•NOTCH信号通路:健康与肝硬化肝组织的单细胞转录组分析证实,NOTCH是关键促纤维化通路;NOTCH1通过调控HSC的转录组变化,促进其活化和纤维化相关基因表达。
•整合素-ILK信号通路:整合素与ECM结合后激活ILK(整合素连接激酶),ILK以组织特异性方式介导HSC的上皮-间质转化(EMT),参与肝纤维化进程;血管紧张素II可通过TGF-β/SMAD信号上调ILK表达,进一步放大纤维化效应。
•JAK/STAT信号通路:JAK/STAT通路被细胞因子(如IL-6、PDGF)激活后,可直接促进HSC活化和肝细胞炎症反应,加速肝纤维化;JAK抑制剂已在动物模型中显示出抗肝纤维化潜力。
•大麻素(CB)信号通路:CB1R信号激活会驱动肝脏氧化应激、炎症和纤维化,在肝纤维化模型中高表达;CB2R则具有抗炎、抗氧化和抗纤维化作用,但在肝纤维化中的具体机制仍存在争议,需进一步研究。
PART 03
逆转“关键点”:纤维化与肝硬化可逆的核心机制
肝纤维化和代偿期肝硬化的可逆性,核心依赖以下四大机制:
1.肌成纤维细胞凋亡:
通过药物或自身免疫调控,让活化的肌成纤维细胞发生程序性死亡,减少“疤痕制造者”;
2.细胞外基质降解:
肝脏内的基质金属蛋白酶(MMPs)能降解胶原蛋白等细胞外基质,而TIMPs会抑制MMPs的活性。纤维化逆转时,MMPs表达升高,TIMPs表达降低,实现疤痕组织的降解;
3.HSC失活:
部分活化的HSC可逆转回静息态,停止分泌细胞外基质;
4.免疫细胞调控:
自然杀伤(NK)细胞能通过分泌IFN-γ和表达TRAIL(肿瘤坏死因子相关凋亡诱导配体),直接杀伤活化的HSC,同时抑制炎症反应,促进纤维化逆转。有研究表明,巨噬细胞的表型极化是纤维化逆转的重要环节,M2型巨噬细胞向M1型极化后,可通过吞噬活化的成纤维细胞或分泌抗纤维化因子,抑制纤维化进展。
对于肝硬化而言,逆转的核心是“病因根除+长期修复”——只有彻底消除病毒、酒精等损伤因素,再通过靶向药物持续抑制纤维化通路,才能逐步减少假小叶数量、改善肝脏结构。但肝硬化的逆转程度有限,无法完全恢复正常肝脏架构,因此早期干预仍是重中之重。
综上,肝纤维化的发病机制是多细胞协同、多通路交织的复杂网络,核心在于肝星状细胞的活化与细胞外基质的过度沉积。
明确关键细胞与信号通路的作用,是创新药物研发的核心靶点;而早期干预、根除病因则是逆转纤维化的关键。
后续我们将持续解析肝纤维化创新药的研发突破,敬请关注系列第三期内容。
💡关注“小牛医药”,不错过每一次精彩的知识分享!
参考文献
1、Di X, Li Y, Wei J, Li T, Liao B. Targeting Fibrosis: From Molecular Mechanisms to Advanced Therapies. Adv Sci (Weinh). 2025 Jan;12(3):e2410416.
2、Vandyck K, McGowan DC, Deval J, et al. Discovery and Preclinical Profile of ALG-055009, a Potent and Selective Thyroid Hormone Receptor Beta (THR-β) Agonist for the Treatment of MASH. J Med Chem. 2024 Sep 12;67(17):14840-14851.
3、Wang H, Zhang J, Chai J, et al. Fluorofenidone ameliorates cholestasis and fibrosis by inhibiting hepatic Erk/-Egr-1 signaling and Tgfβ1/Smad pathway in mice. Biochim Biophys Acta Mol Basis Dis. 2022 Dec 1;1868(12):166556.
编辑:陈福鼎
审核:陈亚红
润色排版:周安楠