
2025年9月28日、2026年1月16日,国务院总理李强先后签署第818号令《生物医学新技术临床研究和临床转化应用管理条例》(2026年5月1日施行)和第828号令《中华人民共和国药品管理法实施条例》(2026年5月15日施行),两令在前后半月内相继执行,标志着我国生物医药监管体系迎来深刻变革,”双轨制”监管体系的正式成型——”技术”归国家卫生健康委员会(卫健委),”药品”归国家药品监督管理局(药监局),从根本上厘清长期监管模糊地带。
本文将从新旧政变更、政策深意及药企行动指南三个维度,为读者梳理两部法规的核心要点。

PART 01
818号令:生物医学新技术监管首部“国字号”法规
818号令共7章58条,在我国境内从事生物医学新技术临床研究、临床转化应用及其监督管理,适用《条例》的规定。
1.适用范围界定
- 本条例所称生物医学新技术,是指以对健康状态作出判断或者预防治疗疾病、促进健康为目的,运用生物学原理,作用于人体细胞、分子水平,在我国境内尚未应用于临床的医学专业手段和措施。(第三条)
- 政策深意:这与2022年《药品管理法实施条例(征求意见稿)》中有关CGT(细胞与基因治疗)产品的分类思路一脉相承,同时首次以行政法规形式明确细胞治疗、基因治疗、组织工程、脑机接口等前沿技术的监管属性,填补法律空白。
2.备案+审批双轨制管理
- 818号令:确立”临床研究备案+转化应用审批”新路径,为技术转化提供合法通道。
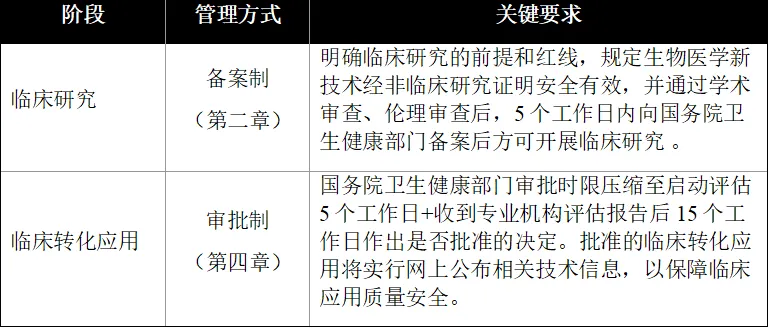
- 与旧政对比:
- 2015年前:细胞治疗作为第三类医疗技术,存在临床应用准入审批。
- 2015-2025年:魏则西事件后,第三类医疗技术准入审批取消,干细胞、免疫细胞治疗仅可开展科研性质临床研究,不得收费、不得转化应用。
3.机构门槛硬性约束(第十一条)
- 818号令:临床研究机构必须为三级甲等医疗机构,且具备:
- 学术委员会、伦理委员会
- 项目负责人具备执业医师资格+高级职称,并以临床研究机构为主要执业机构
- 保存记录30年(涉及子代的永久保存)。
- 产业影响:非三甲医院及美容机构、养生馆等彻底退出市场,行业资源向头部医院集中
4.临床转化应用(商业化路径)
- 收费许可:临床研究阶段严禁收费,医疗机构仅在获得批准的临床转化应用技术,可以按照规定收取费用(第三十四条)。
- 优先审批: 针对严重危及生命且无有效手段的疾病,予以优先审查审批(第三十二条)。
- 公开透明: 批准的技术需公布名称、机构条件、操作规范(第三十三条)。
- 紧急应用: 应对突发公卫事件,可紧急应用正在研究中的技术(第三十六条)。
- 动态评估与暂停: 安全性有效性认识改变时,需再评估,期间暂停临床应用(第三十七条)。
5.受试者权益保护升级
- 知情同意:实施临床研究,应当取得受试者或者其监护人的书面知情同意;变更临床研究方案对受试者权益可能产生影响的,需要重新取得知情同意。
- 权益保障:开展临床研究不得向受试者收取与临床研究有关的费用;临床研究造成受试者健康损害的,临床研究机构应当及时予以治疗;鼓励临床研究购买商业保险,为受试者提供更多保障;相关机构依法保护受试者的个人隐私、个人信息。
6.其他处罚力度显著加大,可查封场所、扣押设备,最高罚款1000万,终身禁业;明确举报奖励与保护,引入“吹哨人”制度,内部违规被曝光风险增加。
PART 02
828号令:药品全生命周期管理“底层逻辑重构”
828号令共9章89条,聚焦药品全生命周期管理,强化监管红线,释放鼓励创新,促进药品产业高质量发展,有针对性地细化补充制度措施。
1.创新激励制度体系化
- 市场独占期激励:
- 儿童药:新品种、新剂型、新规格、增加适应症,符合条件给予不超过 2 年市场独占期;
- 罕见病药:承诺保障供应的,给予不超过 7 年市场独占期;
- 惩罚机制: 持有人不履行供应承诺,独占期终止。
- 数据保护制度:
- 保护期限: 自行取得且未披露的试验数据,保护期自注册之日起不超过 6 年。
- 保护内容: 保护期内,他人未经许可使用该数据申请注册不予许可(除非自行取得数据)。
- 例外: 公共利益需要或已采取措施防止不正当商业利用时可披露。
2.MAH责任压实与分段生产突破
- 828号令:”药品上市许可持有人确有需要的,可以委托符合条件的药品生产企业分段生产:(一)生产工艺、设施设备有特殊要求的创新药;(二)临床急需的药品、应对突发公共卫生事件急需的药品或者储备需要的药品;(三)国务院药品监督管理部门规定的其他药品。
- 828号令:药品注册标准应当符合《中华人民共和国药典》的通用技术要求,并且不得低于相应的国家药品标准。强制MAH持有人应当根据国家药品标准变化,及时修订,提出质量变更申请。
- 政策深意:MAH必须建立”覆盖药品生产全过程和全部生产场地的统一质量保证体系”,需持续监控药典修订,委托不转嫁责任。
3.其他核心变化
- 原料药管理:允许转让原料药批准证书,转让申请自药监部门受理申请之日起20个工作日内进行审查并作出决定。明确原料药颁发批准证书且有效期为 5 年,届满需申请再注册。
- 生产前置合法化:”在取得药品批准证明文件前生产且通过相应药品生产质量管理规范符合性检查的商业规模批次产品,符合药品上市放行要求的,可以在取得药品注册批准文件后上市销售。
PART 03
政策深意:双轨协同监管,推动产业高质量发展
两项法规同步落地并非巧合,而是国家在生物医药领域的系统性制度安排,其政策深意可归纳为以下四点:
1.确立“双轨制”监管架构,理清技术与产品的监管边界。818号令由卫健委主导监管,适用于难以标准化的治疗技术;828号令由药监局主导监管,适用于可标准化批量生产的药品。这一架构明确了各自的监管主体和职责,从根本上解决了过去CGT领域“既像药品又像技术”的监管困境。
2.以临床价值为导向,实现“清场”与“加速”并举。
818号令通过机构准入严控、禁止外包原则等,淘汰游走于灰色地带的违规主体,美容院、养生馆等非医疗机构彻底出局,杜绝 “伪创新”“乱收费”,为合规技术转化扫清障碍,净化行业生态环境;828号令则通过加快上市注册机制和创新保护制度,为真正具有临床价值的创新药开辟快速通道。
3.压实MAH主体责任,推动行业优胜劣汰。 828号令将MAH确立为药品安全的“唯一最终责任人”,责任不可外包。这一原则将显著提升行业准入门槛,促使药企在选择CRO、CDMO等服务商时从“价格导向”转向“质量与合规导向”。
4.全链条控制风险,保护患者权益。818号令明确临床研究免费、转化应用审批制,避免患者被虚假技术误导,保障医疗技术的公益性与安全性;828号令从注册、生产、经营到使用,全环节压实主体责任,尤其是分段生产、委托生产、网络销售等易风险环节,提升药品质量可控性。
5.构建全生命周期监管闭环。 从818号令的临床研究备案管理,到828号令的药品研制、注册、生产、经营、上市后监管全链条覆盖,形成了从“实验室—临床研究—转化应用/药品注册—上市后监管”的完整监管闭环。
PART 04
新政下药企业行动建议
1.重新评估CRO合作伙伴选择标准。
828号令强化了MAH的“唯一最终责任人”地位,企业选择CRO时必须将重心转向数据真实性与流程合规保障。重点考察CRO数据全生命周期可追溯、防篡改的能力,严格审查其在伦理审查、受试者保护等关键流程上的标准操作和历史记录。
2.关注市场独占期窗口,优化产品管线优先级。
罕见病用药最长7年、儿童用药最长2年的市场独占期,以及最长6年的试验数据保护期,将深刻影响产品估值和竞争格局。建议企业在管线布局时,优先评估产品是否符合上述激励条件,并建立独占期到期前的仿制药预警机制。
3.建立全生命周期数据管理体系。
建议企业提前布局符合GCP和GLP要求的数据管理系统,建立从非临床研究到临床试验再到上市后研究的数据闭环;生产端严控质量,规范委托与分段生产;经营端规范网络销售,严格执行禁售清单,网络交易第三方平台需建立质量管理体系;合规端强化风险防控,杜绝数据造假。新条例第六条要求从事药品研制活动应保证记录和数据“真实、准确、完整和可追溯”。
PART 05
总结
818、828 号令的实施,标志着中国生物医药产业进入 “规范与创新并重” 的新阶段。技术赛道守合规备案,药品赛道抓临床价值与全生命周期合规。建议各企业组织全员进行政策培训,启动内部全流程自查整改,建立合规长效机制,确保平稳过渡,并依托政策红利布局优势赛道,以合规为基础实现高质量发展。
如需深入探讨,欢迎联系小牛医药商务团队:
💡郑总监
联系电话:13758291914(微信同号)
商务邮箱:zym@millibuff.com


