
一、什么是FIH试验,为什么它如此特殊?
FIH试验(First-in-Human),指完成临床前研究后,将研究药物首次应用于人体的试验阶段,其核心任务是在人体中首次验证安全性、耐受性、药代动力学特征,并初步探索药效活性。
这十类药物,需要额外警惕
CDE指导原则及国际监管实践指出,以下十类药物具有更高的内在风险:
1.可能严重干扰生命体征的任何制剂;
2.具有激动剂和拮抗作用的制剂;
3.无研究先例的新型制剂或新作用机制的药物;
4.具有种属特异性、难以进行临床前评估的药物;
5.与自然配体相比具有高效能的制剂;
6.多功能制剂(如双价抗体);
7.以细胞相关为靶点的药物;
8.绕过正常控制机制作用于靶点的药物;
9.以免疫系统为靶点的药物;
10.潜在产生生物放大效应的药物。
二、IND到首例入组:七个风险高发节点
节点一:IND申报安全评估的起点
IND申报是药物进入临床试验的法定门槛。IND申请资料涵盖所有临床前研究数据,是监管机构评估”是否具备进入人体试验条件”的依据。
核心风险:临床前安全性数据包不完整、对高风险特征评估不充分、研究者手册信息滞后,都会影响IND申报质量。
实操建议:IND提交前,组织跨部门(药理、毒理、临床、注册)联合评审,对照十类高风险药物特征进行自评,如有涉及,启动额外安全评估程序。研究者手册在首例受试者入组前须更新至最新版本[ICH GCP E6(R2)第4.4节]。
案例警示:BIA 10-2474事件2016年1月,法国Bial公司研发的FAAH(脂肪酸酰胺水解酶)抑制剂BIA 10-2474,在单剂量递增(SAD)阶段顺利完成后进入多剂量递增(MAD)研究,首名受试者给药后出现严重神经系统损害并最终脑死亡,后续4名受试者也出现不同程度的神经系统毒性。事后调查揭示:同类FAAH抑制剂的研发管线中早已存在神经毒性信号,但这些已知安全信息并未在IND申报阶段得到充分评估与整合;BIA 10-2474以共价结合方式不可逆地抑制FAAH,这一关键药理特性意味着多日给药后药效会持续累积,而临床前毒理评估体系对这一机制风险亦未给予充分重视。
→关键教训:IND申报阶段须系统梳理同靶点/同机制药物的研发历史,特别是失败案例的安全性信息;对共价结合、不可逆抑制等特殊药理机制,临床前毒理评估应设计针对性实验,不能仅依赖常规毒理学框架;高风险机制在IND阶段未被识别,往往在临床中付出更大代价。
节点二:起始剂量选择科学与审慎的平衡
起始剂量的确定是FIH试验最核心的决策之一:剂量过低,可能难以获得有意义的药效探索数据;剂量过高,则直接威胁受试者安全。
NOAEL→HED法(FDA主导):
基于动物毒理学研究,找出最敏感物种中未观察到不良反应剂量(NOAEL),通过异速缩放公式换算为人体等效剂量(HED),再除以安全系数(通常为10)得到人体起始剂量。该方法以毒性终点为核心,适用于传统小分子药物,但对高效能生物制品(尤其是免疫激动剂)存在局限性:依此法计算的起始剂量有时接近甚至超过临床真实的最大耐受剂量(MTD),从而可能高估安全剂量。
MABEL法(EMA优先推荐):
该法源于TGN1412灾难性事件后EMA的倡导。它综合所有体内外非临床数据(包括受体占有率、药效学活性、靶点表达及功能等),通过PK/PD建模计算能够产生最低预期生物效应的剂量。相较于NOAEL法,MABEL更注重药理学效应,对靶向生物制品、激动剂类药物能够提供更审慎的剂量起点,显著降低细胞因子风暴等风险。但需注意,MABEL法也可能导致起始剂量过低,从而增加剂量递增的阶梯数、延缓开发进程。
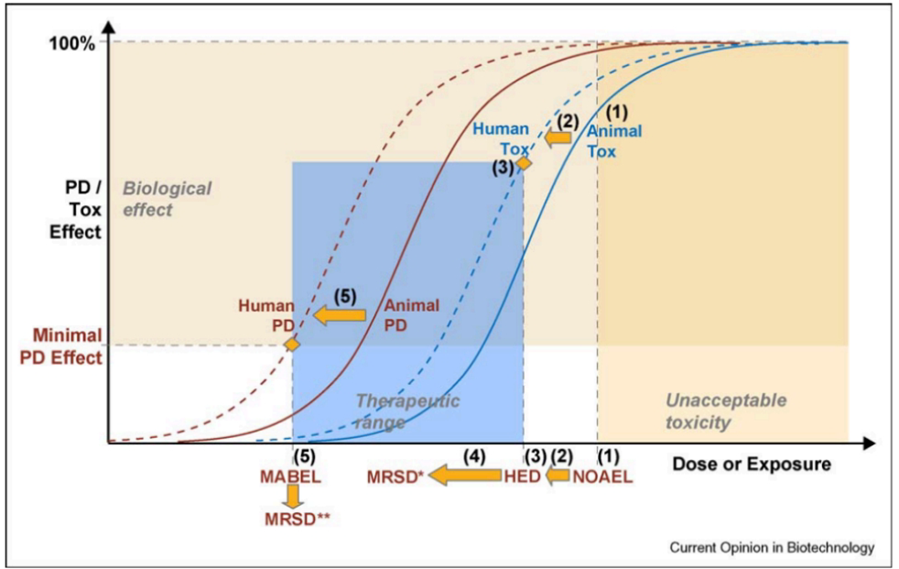
基于MABEL和NOAEL的FIH试验MRSD示意图
最佳实践:
同时计算NOAEL值和MABEL值,取较低者作为起始剂量的参考。这一做法综合了EMA和FDA两大监管框架的核心建议,并经行业实践反复验证,是当前高风险生物制品FIH试验的基本策略。在条件允许时,还可进一步采用基于机制的PK/PD模型来精确推算起始剂量,在安全性与研发效率之间取得更好平衡。
案例警示:TGN1412事件2006年,英国TeGenero公司研发的CD28超级激动剂抗体TGN1412进入I期临床,尽管临床前在猕猴中完成安全性评估,人体给药后数分钟内,6名受试者全部出现危及生命的细胞因子风暴。后续研究揭示:人类CD4+效应记忆T细胞上CD28高表达,与灵长类动物存在物种差异,动物实验数据无法预测人类的极端免疫反应。
→关键教训:高风险生物制剂须优先采用MABEL法;充分评估靶点在人体与动物模型间的表达差异;必要时开展体外免疫毒性测试(如高密度PBMC培养)作为补充验证。
节点三:方案设计与给药策略安全是第一位
方案设计需要回答:如何递增剂量?每次几名受试者?间隔多久?如何设定终止规则?
剂量递增方案的主要类型:
| 方案 | 说明 | 特点 |
| 传统3+3 | 每剂量组纳入3名受试者,若无DLT则进入下一剂量组;若1名出现DLT则扩展至6名进一步评估 | 操作简单,较为保守,但效率较低 |
| i3+3 | 结合贝叶斯统计方法,根据实时数据动态判断最优剂量 | 兼顾安全性与效率,近年应用增加 |
| BOIN | 使用贝叶斯最优区间法,自动判断最优剂量并设置停止边界 | 适合高风险药物,可减少不必要的受试者暴露 |
哨兵给药(sentinel dosing):每个新剂量组中,先由1名受试者给药并观察48-72小时,确认安全后再继续该组其余受试者给药。高风险药物FIH试验的常用保护性设计。
剂量限制性毒性(DLT)定义:DLT定义直接决定试验何时暂停或终止。对于ADC、血液学毒性风险较高的药物,DLT定义应覆盖更广的毒性类型并设置更严格阈值。
特别提示SAD安全≠MAD安全:单剂量递增阶段的安全数据不能自然推演至多剂量递增。对于共价结合型药物、不可逆抑制剂或存在累积毒性风险的药物,MAD方案须额外设置特异性安全监测指标和更严格的停药标准,不可沿用SAD阶段的安全假设。
案例警示:BNT326/YL202事件2024年6月,宜联生物(MediLink)与BioNTech合作开发的HER3 ADC药物BNT326/YL202遭遇FDA部分临床暂停。FDA指出:该药物在较高剂量下可能使受试者面临不合理且显著的患病或伤害风险;临床数据显示存在剂量依赖性的中性粒细胞减少和黏膜炎增加趋势。2024年8月FDA解除暂停,整改措施包括:将后续开发集中于≤3.0mg/kg剂量(该剂量组安全性可控且保留了抗肿瘤活性),修订方案增加TRAE预防和处理规范,更新研究者手册。
→关键教训:ADC等复杂药物在剂量爬坡中可能存在”安全性断崖”在某个剂量阈值以上,风险急剧上升;提前识别这一阈值、设置更严格的DLT定义和暂停规则是关键。BNT326同时说明:发现安全性问题后主动暂停、及时调整方案,是可以继续推进研发的,关键在于响应速度。
节点四:受试者筛选与知情同意伦理的底线
受试者选择的科学考量:选择健康受试者还是患者,须综合考虑药物的预期治疗窗、急性/慢性毒性风险、药物代谢特征以及目标适应症的疾病特点。
知情同意的完整要素(依据《涉及人的生物医学研究伦理审查办法》和《民法典》第1008条):试验目的、流程和潜在风险;替代治疗选择(如有);自愿参与和随时退出的权利;临床试验不得向受试者收费;知情同意书不得包含免除申办方责任的条款。
→关键教训:知情同意书须经伦理委员会正式审查,不得含有免除申办方或研究者责任的条款;获得伦理批准后,方案执行必须严格忠实于批准内容,未经伦理同意不得擅自变更;研究者须定期提交研究进展报告;伦理委员会的跟踪审查职责与初始批准同等重要;临床试验不得向受试者收取任何费用。
节点五:安全性监测与实时信号捕获
四个层次的监测体系:
第一层:临床观察研究者须对受试者进行密切观察,记录所有不良事件(AE),评估其与研究药物的相关性。
第二层:实验室检测根据药物特性设置检测指标和采样时间点,高风险药物须在给药后设置更密集的采样节点。
第三层:SUSAR快速报告致死或危及生命的SUSAR须在7天内报告(部分情况24小时内),其他严重SUSAR须在15天内报告(依据ICH E2A指导原则及GCP法规要求)。
第四层:独立数据监查委员会(iDMC)高风险FIH试验应在启动前组建iDMC,制定章程,明确iDMC有权建议试验暂停或终止。
节点六:伦理审查与监管沟通持续的责任
伦理审查不是”一次性”动作,而是一个持续过程。”批了就不管”的”批而不审”现象,是严重违反伦理委员会职责的行为。伦理委员会须对已批准试验进行持续跟踪审查,研究者须定期提交研究进展报告;当试验过程中出现安全性信号时,申办方应主动与监管机构沟通。
案例警示:苏州罗某细胞治疗致死案(2025年判决)苏州某医院开展的细胞治疗临床研究中,知情同意书未经伦理委员会正式审查,且含有违规的免责条款;受试者入组后,医院并未按伦理批准方案实施治疗;研究者未提交研究进展报告;伦理委员会亦未履行跟踪审查职责。受试者最终死亡,法院判决试验公司和医院共同承担70%责任,赔偿90余万元。
→关键教训:伦理审查须贯穿试验全程;研究者须定期提交研究进展报告,确保伦理委员会持续知情;任何实质性方案变更均须重新提交伦理审查;伦理委员会跟踪审查失职与初始批准不当一样,可能造成不可挽回的后果。
节点七:申办方内部监管体系守好第一道门
伦理委员会和监管机构是外部防线,而申办方/CRO自身的内部治理体系,是受试者安全的”第一道门”。即便外部审批全部到位,如果申办方内部管理失序,安全风险仍可能在不为人知的角落积累。
申办方内部的五大核心职责:制定并严格执行FIH试验安全风险管理计划(RMP),明确识别、评估和管控风险的策略;建立完善的临床研究药品全链条管控体系,包括专人专库管理、发放记录与可追溯机制;制定并严格执行SOP,确保规程不只存在于纸面;建立内部质量管理体系(QMS),定期开展自查与稽查,及时识别并纠正偏差;构建安全信号快速响应机制,确保问题从发现到上报到处置的每个环节都有明确责任人和时限。
案例警示:重庆“卡度尼利“临床研究药品外流事件(2025年)2025年7月,重庆市药监局通报:康方药业卡度尼利单抗的临床研究药品,遭销售人员通过伪造医院研究立项文件及伦理批件的手段套取后流入市场,涉及销售人员伪造文件、医生违规推荐、药店违规发放、诊所违规输注等多个环节。这一事件并非单一环节失误所致,而是申办方对临床研究药品的发放管控链条存在系统性漏洞,使得伪造文件得以一路畅通。
→关键教训:临床研究药品须专人专库管理,建立完整发放和追溯记录,申办方须对药品流向实施实质性监控而非仅依赖形式登记;对商业推广人员接触临床研究药品须有明确禁区规则;每一个合规环节的疏漏都可能在链条最薄弱处引发系统性风险。
案例警示:康龙化成实验室窒息事故(2025年)2025年6月3日,北京某实验室2名操作人员在未按公司安全规程的情况下进行隔离器操作,导致缺氧窒息,双双死亡。事故调查后,企业总裁被建议罚款40%年薪。这一事故虽发生于临床前研究阶段,但深刻揭示了安全操作规程执行的系统性缺陷规程的存在不等于规程被执行,培训到位不等于习惯养成。
→关键教训:安全操作规程是最后防线,不得以任何理由绕过;须定期开展安全培训和应急演练,并保留培训记录;对违规行为建立问责机制;合规意识须从管理层向一线操作人员全面渗透,否则风险只是被转移,而非被消除。
三、执行检查清单:七个节点,一份可复用的自检工具
以下清单供IND到首例入组全流程自查参考。
IND申报阶段(参考:CDE FIH安全风险管理计划指导原则、CDE肿瘤药扩展队列研究指导原则、FDA CDER FIH临床考量指南)
£临床前安全性数据包是否完整?药效学、药代动力学和毒理学数据是否齐全?
£是否对照十类高风险药物特征进行自评?如有涉及,是否提交了额外安全评估?
£是否系统梳理了同靶点/同机制药物的研发历史,包含失败案例的安全性信息?
£对于高风险生物制品,是否提供了MABEL法计算依据?
£研究者手册在首例受试者入组前是否已更新至最新版本?
起始剂量选择阶段(参考:FDA起始剂量指南、EMA FIH指南、FDA PBPK分析指南)
£是否确定采用NOAEL→HED还是MABEL?选择的科学依据是否充分?
£是否同时计算了NOAEL和MABEL,取较低值作为参考?
£是否评估了靶点在人体与动物模型间的物种差异?
£条件允许时,是否考虑了基于机制的PK/PD模型或PBPK模型对起始剂量的进一步验证
方案设计阶段(参考:CDE扩展队列研究指导原则、EMA FIH指南、CDE FIH安全风险管理计划指导原则)
£是否评估了靶点在人体与动物模型间的物种差异?
£剂量递增方案(3+3/i3+3/BOIN)的选择是否与药物风险匹配?
£DLT定义是否覆盖了药物的已知毒性特征?是否设置了更严格的阈值?
£对于共价结合药物或ADC,是否额外评估了多剂量累积风险,并在MAD方案中设置了特异性安全指标?
£终止标准是否清晰可执行?
£是否规定了安全性信号的审核与上报流程?
伦理与知情同意阶段(参考:《药物临床试验质量管理规范》、《涉及人的生物医学研究伦理审查办法》、《民法典》第1008条)
£是否已确定受试者人群选择(健康志愿者vs患者)并说明科学依据?
£知情同意书是否经伦理委员会正式审查?
£知情同意书是否包含免除申办方责任的条款?(须为”否”)
£是否告知了受试者费用承担?(临床试验不得收费)
£方案执行是否严格遵循伦理批准版本?任何实质性变更是否已重新提交伦理审查?
安全性监测阶段(参考:ICH E2A不良事件快速报告标准、《药物临床试验质量管理规范》、CDE SAE相关性评价指导原则)
£实验室检测指标和采样时间点是否充分覆盖药物的已知毒性?
£是否组建了iDMC并制定了章程?章程中是否明确iDMC有权建议暂停或终止试验?
£SUSAR报告时限(7天/15天/24小时)是否清晰传达至所有相关方?
£是否建立了安全性信号的快速内部升级机制?
伦理审查与监管沟通阶段(参考:《药物临床试验质量管理规范》、《涉及人的生物医学研究伦理审查办法》)
£是否建立了伦理跟踪审查机制?
£研究者是否按要求定期提交研究进展报告?
£是否建立了与监管机构的主动沟通机制?出现安全性信号时是否已主动报告?
申办方内部监管体系阶段(参考:《药物临床试验质量管理规范》、CFDIⅠ期临床试验管理指导原则、CDE FIH安全风险管理计划指导原则)
£是否制定了FIH试验安全风险管理计划(RMP)?RMP中的风险识别和管控策略是否已落实到具体执行层面?
£临床研究药品是否实行专人专库管理?发放记录和可追溯机制是否完整?对商业推广人员接触临床研究药品是否有明确禁区规则?
£SOP体系是否覆盖所有关键操作流程?规程是否仅存在于纸面?
£是否建立了内部质量管理体系(QMS),定期开展自查与稽查?
£是否对关键文件(伦理批件、研究者手册等)建立了版本控制和发放记录?
£是否构建了安全信号的快速响应和内部升级机制?
结语
从IND批件到首例受试者入组,这段路程上的每一个节点IND申报、起始剂量选择、方案设计、知情同意、安全性监测、伦理合规、申办方内部监管都在考验着团队的专业能力与责任意识。
TGN1412事件让我们认识到物种差异的不可预测性,BIA 10-2474告诉我们SAD安全不等于MAD安全,BNT326案例说明剂量爬坡中可能存在意想不到的安全断崖。国内的苏州罗某案、重庆卡度尼利事件和康龙化成事故,则从不同维度提醒我们:合规不是形式,而是受试者安全的制度保障。
我司在FIH试验的风险评估与申报策略、方案设计与给药策略制定、伦理合规体系构建及全流程质量控制等领域积累了丰富经验,可为创新药申办方提供从IND申报到首例入组的全流程专业支持。欢迎联系商务团队探讨合作。
参考文献
[1]U.S.Food and Drug Administration.Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers[S].Rockville:FDA,2005.
[2]International Conference on Harmonisation.Clinical Safety Data Management:Definitions and Standards for Expedited Reporting(E2A)[S].Geneva:ICH,1994.
[3]U.S.Food and Drug Administration.CDER’s Clinical Consideration for First-in-Human Trials[EB/OL].(2024-12-10)[2025-04-28]. https://www.fda.gov/media/184323/download.
[4]European Medicines Agency.Guideline on Strategies to Identify and Mitigate Risks for First-in-Human and Early Clinical Trials with Investigational Medicinal Products(Revision 1)[S].London:EMA,2007.
[5]国家药品监督管理局药品审评中心.肿瘤药首次人体试验扩展队列研究技术指导原则(试行)[S].北京:CDE,2012.
[6]国家药品监督管理局药品审评中心.药物首次人体试验安全风险管理计划指导原则[S].北京:CDE,2024.
[7]Suntharalingam G,Perry M R,Ward S,et al.Cytokine Storm in a Phase 1 Trial of the Anti-CD28 Superagonist TGN1412[J].N Engl J Med,2006,355(10):1018-1028.
[8]Hünig T.The storm has cleared:lessons from the CD28 superagonist TGN1412 trial[J].Nat Rev Immunol,2012,12(5):329.
[9]Eastwood D,Findlay L,Bird C,et al.Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4+effector memory T-cells[J].Br J Pharmacol,2010,161(3):512-526.
[10]陈霞.BIA 10-2474药物临床研究中5例健康受试者神经系统损害及死亡事件的启示[J].协和医学杂志,2018,9(3):256-260.
[11]重庆市药监局.关于康方药业临床研究药品外流事件调查通报[R].重庆:重庆市药监局,2025-07-13.
[12]苏州市中级人民法院.癌症患者参与临床医疗试验项目时病故试验公司和医院被判担责[N].人民法院报,2025-12-09.
[13]北京经济技术开发区应急管理局.康龙化成”6.3″隔离器内窒息死亡事故调查报告[R].北京,2025-09.
[14]BioNTech SE.Lift of Partial Clinical Hold for BNT326/YL202[EB/OL].(2024-08-19)[2025-05-08].https://investors.biontech.de/news-releases/news-release-details/lift-partial-clinical-hold-bnt326yl202.
[15]国家药品监督管理局食品药品审核查验中心.药物Ⅰ期临床试验管理指导原则[S].北京:CFDI,2025.
[16]凯莱英临床(凯诺).首次人体研究中MABEL的应用场景[EB/OL].(2024-08-01)[2026-05-08].https://www.clin-nov.com.cn/xwzx/html/609.html.
[17]U.S.Food and Drug Administration.Physiologically Based Pharmacokinetic AnalysesFormat and Content Guidance for Industry[S].Silver Spring:FDA,2018.
[18]国家药品监督管理局.药物临床试验质量管理规范[S].北京:NMPA,2020.
撰稿:李晓宇
审核:陈亚红
排版:吴华芳


