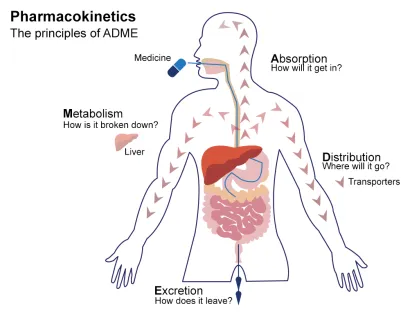
基于生理学的药代动力学模型(physiologically based pharmacokinetic,PBPK模型)是一种机制驱动的数学模型,通过整合解剖结构、生物化学过程及物理化学原理,定量描述药物在体内的吸收、分布、代谢与排泄(ADME)行为。该模型将机体划分为具有明确生理意义的器官或组织隔室(如肝脏、肾脏、肠道等),各隔室通过血液循环动态连接,并基于真实生理参数构建——包括组织容积、血流速率、代谢酶/转运体表达量、组织-血浆分配系数等关键生物学指标,从而实现对药物体内过程的精确模拟。
CDE于2024年发布《模型引导的创新药物剂量探索和优化技术指导原则》,模型引导的药物研发通过采用建模与模拟(Modelling&Simulation, M&S)技术对生理学、药理学以及疾病过程等信息进行整合和定量研究,从而指导新药研发和决策。M&S已应用于药物研发的多个阶段,可在药物研发的多个关键决策点发挥重要作用。
PBPK在新药研发过程中的作用
在临床前阶段,PBPK分析整合稀疏数据,与非房室模型互补,评估实验动物体内药动学行为、PK/PD及安全性特征。它支持首次人体试验(FIH)起始剂量、递增方案设计及最大耐受剂量预测,并有助于确定药物靶标和理解作用机制。
针对健康人群的Ⅰ期试验确定安全剂量、最大耐受剂量及药动学特征。PBPK可深度挖掘数据价值,揭示食物、性别等因素对药动学的影响,为后期临床给药方案提供指导。
Ⅱ期试验在少量患者中评估安全有效性,是确定策略、适应症和方案的关键阶段。此阶段建立的PBPK与疾病进展定量关系及影响因素,能定量指导后期确证性试验的目标人群选择、给药优化、样本量、采样方案和风险控制。
Ⅲ期在大范围患者中进一步评价适应症、疗效和不良反应。PBPK分析可用于优化给药方案和确定最终剂量(如FDA案例所示),并能成功地将成人研究结果外推至儿童,替代或简化部分儿科试验。
上市后研究在广泛人群中关注特殊人群应用、药物相互作用及罕见不良反应。PBPK建模可识别有临床意义的影响因素(年龄、体重、肝肾功能、合并用药、基础疾病等),支持特殊人群剂量选择,并用于基于模型的荟萃分析比较产品疗效。
药物研发中的PBPK建模策略
PBPK模型的建立主要涉及两个部分:模型参数的选择和模型结构的建立。模型参数包括生理相关参数和药物相关参数。PBPK建模通常采用“自下而上”的方法,建立PBPK模型一般遵循如下步骤:
① 收集参数
生理相关参数包括人类和临床前物种的生理参数(如组织体积、血流量、肾小球滤过率、每克肝脏微粒体蛋白/肝细胞数量、血浆蛋白、酶和转运蛋白丰度等),上述数据可在已发表的文献中获得,同时也内置在 GastroPlus、PKSIM 等 PBPK 软件内。
药物相关参数包括理化性质(分子量、pKa、药物的碱性或酸性)、溶解度(logD)和渗透性、血细胞和血浆蛋白结合(如血浆中未结合的分数(fup)、血浆分配(B: P))、转运体对药物分布的影响,以及肝脏或肝外酶代谢的体外数据等(如内在清除率(CLint))。
② 确定模型结构
根据实验目的和药物在人或动物体内的分布情况,选择要纳入模型的相关器官或组织。模型结构通常包括身体核心组织/器官,如血室(动脉和静脉)、代谢消除器官(通常是肝脏和肾脏)和脂肪组织,也包括给药部位(如肺和肠道)、药物作用的器官(如大脑和心脏)和对质量平衡有重要影响的器官(皮肤、骨骼和肌肉等)。
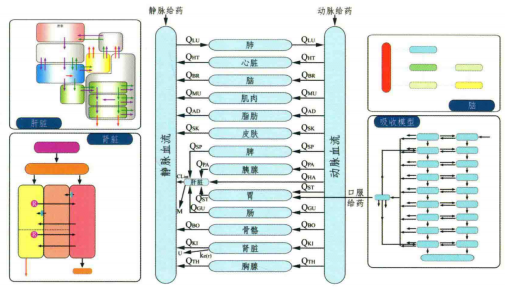
图1 PBPK模型示意图
③ 模型运算
用质量平衡微分方程计算各房室内物质变化,这一步通常使用PBPK软件进行。对于非消除器官,可基于血流灌注限速型或膜渗透限速型假设建立微分方程;对于消除组织/器官,还需考虑清除率(CLint)。
④ 模型优化
参数优化:对模型中的参数进行优化,使模型预测结果与实测数据尽可能接近。可通过灵敏度分析确定对模型预测结果影响较大的参数,然后对这些参数进行调整。
不确定性分析:评估模型参数的不确定性对模型预测结果的影响,确定模型的可靠性和稳健性。
⑤ 模型验证
比较实测数据与模型预测结果,考察模型预测的准确性,确定模型的应用范围是否合理。一个 PBPK模型建立并被验证后,可用于药代动力学外推,不仅可用于对健康人的药代动力学外推,也可用于给药途径之间的外推、不同给药剂量之间的外推、不同物种之间的外推等。
核心应用场景
场景一:首次人体试验(FIH)剂量预测
基于TGN1412事件的重大教训(2006年),EMA于2007年率先建立首次人体试验(FIH)的监管框架,要求系统性识别与防控创新药早期临床风险。FIH聚焦于首次在人类受试者中评估药物安全耐受性,其核心挑战在于通过剂量递增策略探索最大耐受剂量(MTD),这一过程因缺乏人体数据而隐含显著不确定性,需通过机制化建模(如PBPK)和严格风险管理策略降低危害概率。
PBPK模型在首次人体试验(FIH)剂量预测中通过整合生理机制和药物特性,实现从临床前数据向人体的精准外推,其建模流程可参考:
① 临床前数据整合:收集药物理化性质(logP、pKa、溶解度)、蛋白结合率、体外代谢酶动力学(CYP酶Km/Vmax)、渗透性(Caco-2)及动物PK数据等。
② 种属外推与人体参数预测:将动物药物动力学行为机制性外推至人体。
③ 模型构建与验证:基于人体生理结构建立多房室模型,用动物PK数据验证模型可靠性。
④ 安全边界计算:结合NOAEL(无观察不良反应剂量) 和动物暴露量,预测人体安全起始剂量(MRSD)。
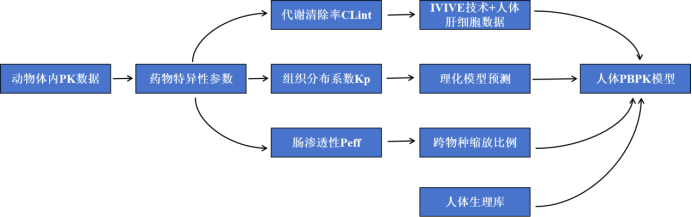
图2 从动物PK到人体PBPK模型的参数转化路径
场景二:药物相互作用(DDI)风险评估
PBPK模型可以通过整合药物性质、系统/生理等信息,并结合临床数据,有效预测药物体内过程,进而考察疾病影响、特殊人群、转运蛋白介导的药物相互作用(DDI)等未知用药场景,为精准治疗提供剂量建议。
PBPK模型在DDI评估中的关键应用如下:
表1 DDI核心应用场景
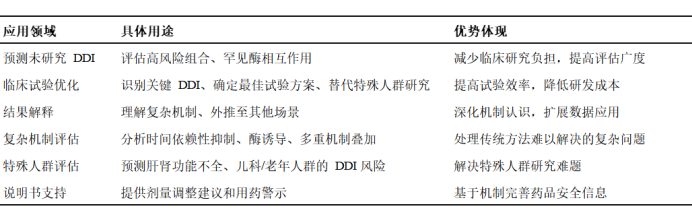
场景三:特殊人群用药优化
覆盖人群:肝肾功能不全、儿童、孕妇、肥胖人群等
如儿童PBPK模型是一个从成人PBPK模型外推的过程。因此,为了使这个外推过程成立,需要人为地规定一些假设:
① 药物在儿童以及成人体内的清除途径相同,差异仅体现在清除率大小上;
② 假设儿童的PBPK模型结构与成人PBPK模型结构一致,这样才可以进行有效的外推;
③ 疾病状态下的儿童与正常儿童相比,发育水平不受影响(除去目前已知对儿童发育有影响的疾病);
④ 除了与年龄相关的药物/系统特异性参数,其余参数在成人和儿童PBPK模型中保持不变。儿童PBPK模型建立的一般先建立该药物成人的PBPK模型并验证,然后再通过改变年龄相关的系统参数,从而建立药物在儿童体内的PBPK模型。
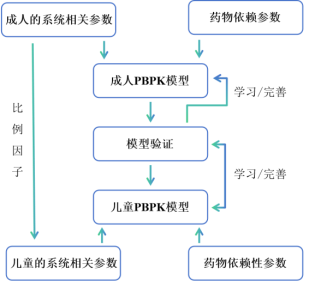
图3 儿童PBPK模型构建的机制框架
场景四:生物等效性豁免
生物等效性豁免是指对影响药物生物等效性的物理、化学及生物性质等因素已有足够了解,能够合理推理出其体内的生物等效性,没有必要通过实际BE试验加以证明,这类药物可豁免人体生物等效性试验。
生物药剂学分类系统(biopharmaceutics classification system,BCS)最早是由Amidon在1995年提出的基于药物的溶解性和渗透性进行药物分类的一种科学系统,根据药物溶解性和肠道渗透性将药物分为四类。BCS分类特点及其代表药物如图:
表2 BCS分类特点及代表药物

目前,国际上主要药品监管机构均发布了基于BCS的生物等效性豁免相关指导原则,我国原国家食品药品监督管理总局也于2016年5月发布了我国基于BCS分类的《人体生物等效性试验豁免指导原则》。虽然具体技术要求并不完全相同,但大多都允许BCSI类和BCSIII类药物的BE豁免申请,且其PBPK模型也都较为成熟。目前国内外暂时均未见关于BCSII类和BCSIV类药物的生物等效豁免的指导意见,但应用PBPK模型对这类药物的体内行为进行预测,以探索分析其可能的豁免标准的研究有很多。
编辑:聂茹佳
审核:宫雪
批准:陈亚红
声明:部分内容和图片源自网络,如侵权可联系删除或注明。